Clinical Implications of New Insights into Hepcidin-Mediated Regulation of Iron Absorption and Metabolism
Focus
Key insights
A deeper understanding of the pathways involved in iron homeostasis have overthrown the old paradigm that the human gut is poorly designed for iron absorption. Instead, the normal physiological status in humans is one where iron absorption is constantly suppressed in order to maintain stable iron levels in the absence of active mechanisms for iron excretion. To this end, the central role of hepcidin is to balance the physiological
need for iron against the threat of infection.
Current knowledge
Hepcidin is a hormone that functions as the master regulator of iron homeostasis. It interacts mainly through the cellular iron exporter ferroportin. Ferroportin transports iron from cellular stores (i.e., enterocytes, macrophages, and hepatocytes) into the blood, thereby raising the levels of circulating iron. Hepcidin induces degradation of ferroportin, resulting in a net lowering of iron concentrations. Upregulation of hepcidin even at very low levels of inflammation underscores the importance of curtailing iron levels when there is danger of infection. Addressing iron deficiency in poor populations therefore needs to combine hygiene and infection control alongside iron supplementation in order to be safe and effective.
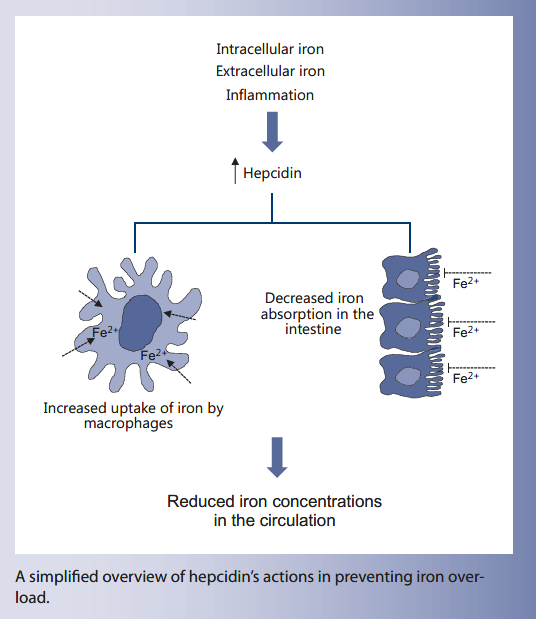
Practical implications
Hepcidin, ferroportin, and their regulators are potential targets for the treatment of iron disorders and anemias. Iron overload could be modulated by artificially enhancing hepcidin. To this end, mini-hepcidins that can perpetuate hepcidin action are being
explored. Conversely, the use of hepcidin antagonists could be useful for the treatment of anemia related to chronic disease or cancer chemotherapy. However, when designing interventions that affect iron levels, great care should be taken not to exceed
safe iron thresholds.
Recommended reading
Atkinson SH, Armitage AE, Khandwala S, Mwangi TW, Uyoga S, Bejon PA, Williams TN, Prentice AM, Drakesmith H: Combinatorial effects of malaria season, iron deficiency, and inflammation determine plasma hepcidin concentration in African children. Blood 2014;123:3221–3229.

Key Insights
A deeper understanding of the pathways involved in iron homeostasis have overthrown the old paradigm that the human gut is poorly designed for iron absorption. Instead, the normal physiological status in humans is one where iron absorption is constantly suppressed in order to maintain stable iron levels in the absence of active mechanisms for iron excretion. To this end, the central role of hepcidin is to balance the physiological need for iron against the threat of infection.
Current Knowledge
Hepcidin is a hormone that functions as the master regulator of iron homeostasis. It interacts mainly through the cellular iron exporter ferroportin. Ferroportin transports iron from cellular stores (i.e., enterocytes, macrophages, and hepatocytes) into the blood, thereby raising the levels of circulating iron. Hepcidin induces degradation of ferroportin, resulting in a net lowering of iron concentrations. Upregulation of hepcidin even at very low levels of inflammation underscores the importance of curtailing iron levels when there is danger of infection. Addressing iron deficiency in poor populations therefore needs to combine hygiene and infection control alongside iron supplementation in order to be safe and effective.
Practical Implications
Hepcidin, ferroportin, and their regulators are potential targets for the treatment of iron disorders and anemias. Iron overload could be modulated by artificially enhancing hepcidin. To this end, mini-hepcidins that can perpetuate hepcidin action are being explored. Conversely, the use of hepcidin antagonists could be useful for the treatment of anemia related to chronic disease or cancer chemotherapy. However, when designing interventions that affect iron levels, great care should be taken not to exceed safe iron thresholds. 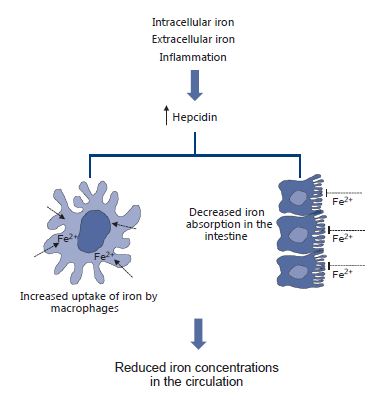
A simplified overview of hepcidin’s actions in preventing iron overload.
Recommended Reading
Atkinson SH, Armitage AE, Khandwala S, Mwangi TW, Uyoga S, Bejon PA, Williams TN, Prentice AM, Drakesmith H: Combinatorial effects of malaria season, iron deficiency, and inflammation determine plasma hepcidin concentration in African children. Blood 2014;123:3221–3229.
Key Messages
-
Iron is a paradoxical nutrient with potential for great benefit but also for causing harm if it is not appropriately chaperoned and regulated.
-
In the past 20 years advances in molecular biology have elucidated many of the mechanisms by which humans tread the delicate balancing act to derive optimal benefit; key amongst these is the discovery of hepcidin which acts as the master regulator of iron.
-
The new insights derived from these basic-science discoveries show that humans exert considerable physiological effort into excluding iron in the face of inflammation and infection, and hence that eliminating these will be at least as important in combatting global iron deficiency as efforts directed towards improving diets.
Keywords
Iron · Hepcidin · Ferroportin · Iron deficiency · Anemia
Abstract
The fact that humans must balance their need for iron against its potential for causing harm has been known for several centuries, but the molecular mechanisms by which we achieve this feat have only been revealed in the last 2 decades. Chief amongst these is the discovery of the master-regulatory liver-derived hormone hepcidin. By switching off ferroportin in enterocytes and macrophages, hepcidin exerts fine control over both iron absorption and its distribution among tissues. Hepcidin expression is downregulated by low iron status and active erythropoiesis and upregulated by iron overload and infection and/or inflammation. The latter mechanism explains the etiology of the anemia of chronic infection. Pharmaceutical companies are actively developing hepcidin agonists and antagonists to combat iron overload and anemia, respectively. In a global health context the discovery of hepcidin shines a new light on the world’s most prevalent micronutrient problem; iron deficiency and its consequent anemia. It is now apparent that humans are not poorly designed to absorb dietary iron, but rather are exerting a tonic downregulation of iron absorption to protect themselves against infection. These new insights suggest that interventions to reduce infections and inflammation will be at least as effective as dietary interventions and that the latter will not succeed without the former.
Introduction
Iron is an intriguing nutrient with many paradoxical characteristics. It is the most abundant element on our planet and yet is hard for living organisms to access due to its very low solubility. Its ability to switch easily between the ferric (Fe3+) and ferrous (Fe2+) state is key to its usefulness in many biological reactions, but managing the electron state also poses a challenge. Iron is an essential and usually highly beneficial nutrient, but at the same time can wreak havoc if it is not appropriately chaperoned to prevent it from causing oxidant damage. In humans these chaperone processes are also critical in withholding iron from bacterial pathogens and maintaining relatively hypoferremic conditions such that microorganisms will not be able to multiply rapidly and can be dealt with by other arms of the innate and adaptive immune systems. This much has been known for many decades with the chemical details of the chaperone mechanisms starting to emerge after Schade’s first descriptions in Science of the iron withholding and bacteriostatic qualities of ovotransferrin and transferrin [1, 2] .
Within the past 2 decades advances in molecular biology methods have revealed the finer details of how iron is absorbed, transported, and utilized. Of special relevance has been the discovery of hepcidin: the master regulator of iron. These insights have transformed our understanding of iron metabolism and have immediate clinical relevance as summarized here.
Twentieth-Century Concepts of Iron Absorption and Regulation
By the 1930s the legendary scientific duo of McCance and Widdowson [3] had concluded that iron homeostasis in humans is controlled by regulating intestinal absorption. Initial attempts to assess the efficiency of intestinal iron absorption from different foods and diets started with fastidious and very time-consuming balance studies [4, 5] . These were superseded by stable isotopic tracer methods which assessed iron utilization (the aggregate outcome of absorption and ultimate utilization for heme production) in the single isotope mode [6] , or true absorption and utilization in the double isotope mode in which one isotope is administered orally and the other intravenously [7].
Arising from these and related studies came the “textbook” knowledge of iron absorption [8] which, simply put, was: (a) that iron absorption is a dynamic process with auto-regulatory feedback (hence, for instance, pregnant women with greater iron needs upregulate their absorption); (b) that heme iron is better absorbed and utilized than non-heme iron; (c) that iron absorption is inhibited by various binding agents in foods (such as phytates and phenolic compounds); (d) that iron absorption is enhanced by co-ingestion of vitamin C; (e) that there is competition for absorption between iron and zinc; and (f) that there is no active process to excrete iron from the body. These were the basic tenets around which most iron interventions were designed and most recommendations were set. Of particular impact was an underlying assumption that the human gut was poorly designed to absorb iron. This prompted extreme practices in which, for instance, young children were (still are) deemed to require 2 mg/kg iron (when their actual daily need is a very small fraction of this), were given highly absorbable iron, often with vitamin C, and dosing was advised to occur between meals to avoid interference by phytates or related “anti-nutrients.” These recommendations for highly nonphysiological dosing regimens have probably caused considerable iatrogenic harm judging from the adverse outcomes of many randomized trials of iron supplementation to children [9] . With the modern insights summarized below, and viewed from a more nuanced understanding of the origins and “intentions” of the finely evolved systems for iron handling, these failures in public health practice are unsurprising. We now know that, far from being ill-designed to absorb iron, children in unhygienic environments are working hard to exclude iron from their systems; and the processes mediating this iron blockade are now well understood.
Molecular Mechanisms of Iron Absorption at the Intestinal Lumen
It seems remarkable that the mammalian divalent cation transporter 1 (DCT1; now termed DMT1) was only discovered and cloned in the rat 20 years ago by Gunshin et al. [10] at Harvard. In the original description in the rat the investigators described the 12 transmembrane loops, its tissue distribution (very high in enterocytes lining the villi of the duodenum but with some expression in most tissues studied), and the fact that mRNA levels were highly upregulated by dietary iron deficiency [10] . They also correctly predicted that the hereditary hemochromatosis gene (HFE) was likely to be part of the iron-sensing regulation of DCT1. 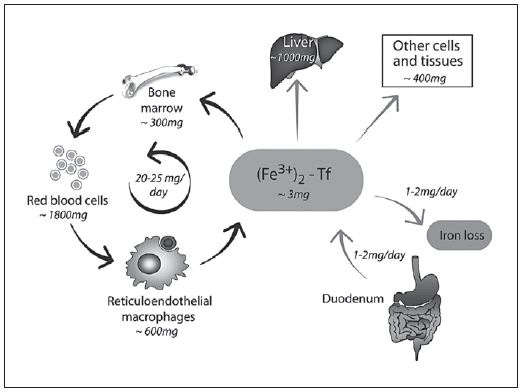
Fig. 1. Iron absorption, recycling, and organ distribution. Values for an adult human male. Reproduced with permission from Hentze et al. [16] . 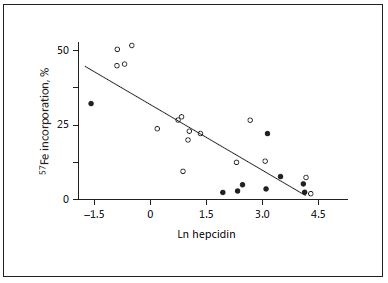
Fig. 2. Hepcidin is the dominant regulator of iron absorption and incorporation into erythrocytes. Data from young Gambian children with postmalarial anemia and control anemic subjects without recent malaria. Reproduced with permission from Doherty and colleagues [22].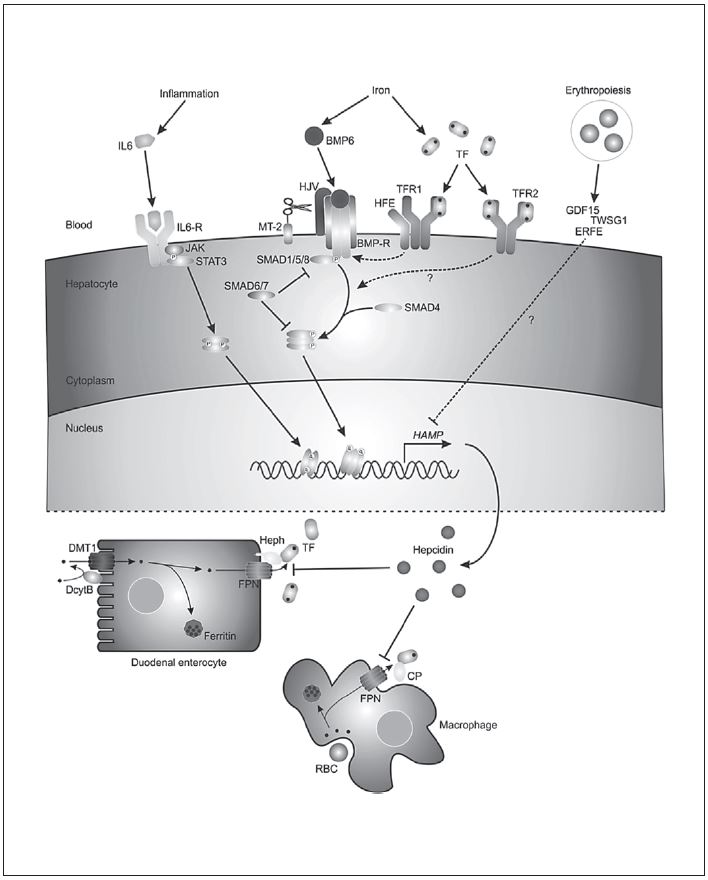
Fig. 3. Current knowledge of the key pathways in the regulation of hepcidin expression. Three main molecular pathways have been identified that modulate hepcidin transcription: JAK/STAT3, BMP/SMAD, and HFE/TfR2 signaling pathway. IL6-induced inflammatory stimuli activate the JAK/STAT3 pathway which in turn triggers hepcidin production. Increased hepatic cellular iron induces BMP6 expression which then interacts with BMPR and HJV, forming a complex, thereby activating the SMAD pathway. The SMAD pathway involves phosphorylation of SMAD1, 5, and 8 (pSMADs), formation of the pSMADs/SMAD4 complex, and subsequent translocation of this complex to the nucleus to activate hepcidin gene expression. Extracellular Tf-Fe2+ mediates a second iron signal. When transferrin saturation increases, Tf-Fe2+ displaces HFE from TfR1. HFE then interacts with TfR2 to form the HFE/ TfR2 complex. Consequently, this complex activates hepcidin transcription via the HJV/BMP/SMAD and/or ERK/MAPK signaling pathways. Furthermore, TMPRSS6 and furin cleave HJV to form a soluble HJV, which can selectively inhibit BMP-induced hepcidin expression. Furin and TMPRSS6 can be regulated by hypoxia and via HIF1. Erythropoietic drive may also control hepci-din expression by EPO production. EPO subsequently stimulates erythroferrone expression, which acts together with TWSG1 to inhibit hepatic hepcidin expression by suppressing the BMP/SMAD pathway. JAK, Janus-associated kinase; STAT, signal transducer and activator of transcription; HFE, human hemochromatosis protein; TfR, transferrin receptor; SMAD, sons of mothers against decapentaplegic; BMP, bone morphogenetic protein; HJV, hemojuvelin; HIF, hypoxia inducible factor; MAPK, mitogen-associated activated protein kinase; ERK, extracellular signal-regulated kinase. Reproduced with permission from Wallace [11] . 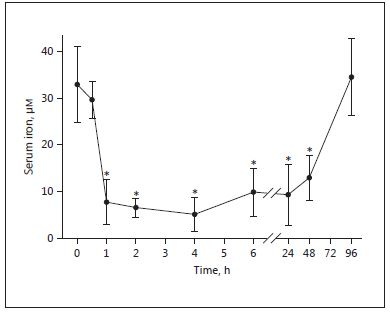
Fig. 4. Hepcidin causes a rapid, sustained fall in serum iron. Mice received a single 50-μg intraperitoneal injection of hHep (human hepcidin). Reproduced with permission from Rivera et al. [26] .
These first discoveries led the way to our current understanding of the molecular mechanism of iron absorption [11] . The absorption of non-heme iron requires that it be reduced to Fe2+ by the action of duodenal cytochrome B (Dcytb) which co-localizes with DMT1 in the brush border membrane [12] . Once inside the enterocyte, iron can either be stored in ferritin (and later lost when the cells are sloughed) or can traverse the cell and enter the circulation via ferroportin. Ferroportin is the only known mammalian iron exporter that facilitates and modulates cellular iron efflux [13] . The exchange of ferrous iron from ferroportin requires its re-oxidation to ferric by the action of ceruloplasmin [14] or (in the intestine) its membrane-bound counterpart hephaestin [15] . Excellent reviews with greater detail are available for readers with a deeper interest [11, 16].
Net Iron Absorption and Iron Recycling
An average adult human loses about 1 mg of iron daily largely through the sloughing of epithelial cells in the intestine. This is a notably small amount when compared to the recommended doses for prevention and treatment of iron deficiency anemia (often 200 times as much or more). In contrast to this very small net intake, the amount of iron internally recycled in the body is considerably more; estimated to be 25–30 mg/day ( Fig. 1 ) [16] . In biology, cycles with a high flux such as this offer the opportunity for exquisite control; if there is a regulatory gateway in the cycle, then closing this gateway has an immediate and profound effect on the concentration of the substrate concerned. As we will see below, this is how the acute phase reaction can elicit such a very rapid hypoferremia. For many years the details of the sensors, regulators, and effectors of these processes remained obscure though it was assumed that there must be one or more hormonal regulators that can transmit information from iron-utilizing end organs (primarily the bone marrow) to the intestine.
Hepcidin: The Master Regulator of Iron Metabolism
Although many pathways and molecular mechanisms are involved in regulating iron metabolism, the current view is that these are all coordinated by the hepatic-derived peptide hepcidin which acts as the master regulator of iron. Hepcidin integrates diverse signals about iron need with counter-regulatory signals designed to suppress iron intake and recycling when there is a threat of infection [17] . The mature 25 amino acid bioactive form of hepcidin contains 8 cysteines linked by 4 disulfide bridges. Hepcidin-like molecules with high homology to human hepcidin are found in mammals, fish, reptiles, and amphibians, but not in birds or invertebrates.
Hepcidin was independently discovered in the early 2000s by 3 groups who were searching variously for novel antimicrobial peptides and for a liver-expressed iron-responsive gene [18–20] . Ganz and colleagues [19] in Los Angeles coined the name hepcidin to describe a hepatic-derived peptide with microbicidal properties. (Note that although the hepcidin molecule does itself possess some antimicrobial activity, this is rather weak compared to peptides such as defensins, and its primary contribution to innate immunity is via regulation of iron.) Experiments manipulating the hepcidin gene Hamp1 soon demonstrated that Hamp1 knockouts became iron loaded and Hamp1 overexpression led to congenitally fatal iron deficiency [17] . Nemeth et al. [21] in the Ganz lab then showed that the molecular mechanism of hepcidin’s action was via binding to, and causing the internalization and degradation of, ferroportin.
Ferroportin is heavily expressed in 2 sites (on the basolateral membrane of enterocytes and on macrophages) and thereby regulates both iron absorption and tissue redistribution [17, 21] ( Fig. 1 ). Hepcidin-mediated down-regulation of ferroportin on the enterocytes blocks iron egress causing a build-up of intracellular iron which in turn blocks further uptake by DMT1. Note that by exerting its control on the basolateral side of the cell it also blocks the uptake of iron released from heme by the action of heme oxygenase. Figure 2 shows that iron utilization by young anemic children is very efficient at low levels of hepcidin but is almost entirely blocked when hepcidin levels are high [22]. Ferroportin blockade in macrophages prevents iron recycling [21] . 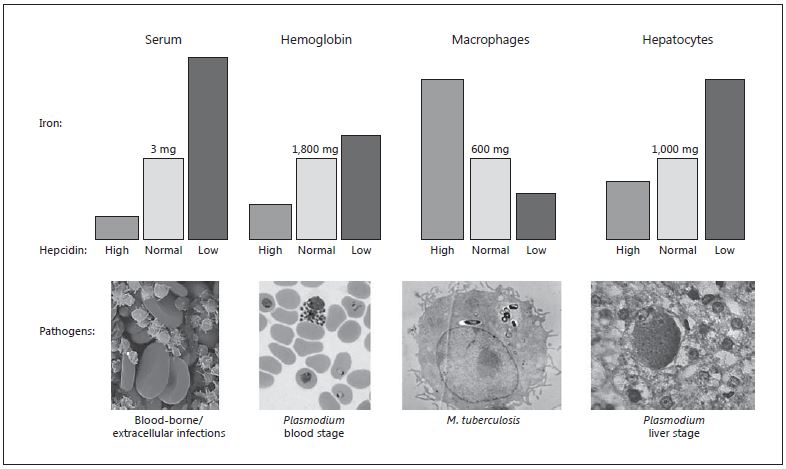
Fig. 5. Hepcidin, iron partitioning, and infectious pathogens. By inhibiting ferroportin, hepcidin controls the total amount of iron entering the circulation as well as the compartments and cell types to which the iron is directed. The usual amounts of iron in serum (bound to transferrin), hemoglobin, macrophages, and hepatocytes are given (represented by light gray columns). In conditions of high hepcidin (dark gray columns), iron recycling through macrophages is inhibited while consumption of iron by the bone marrow continues, leading to a rapid decline in serum iron levels. Over time, persistently high hepcidin reduces iron absorption and locks iron in the reticuloendothelial system, so that hemoglobin iron levels wane, causing anemia. Iron redistribution also leads to less iron in hepatocytes, which express low levels of ferroportin. Conversely, when hepcidin is low (black columns), iron is highly absorbed and readily released from macrophages, leading to elevated levels in serum and moderately increased hemoglobin. The excess iron in the system accumulates in parenchymal tissues, most frequently in he-patocytes. Note that the changes in iron levels represented by the columns are intended to represent trends rather than quantitative measures. Iron-requiring pathogens often inhabit specific anatomical compartments or target particular cell types. Examples include some types of bacteria that thrive and become pathogenic in the bloodstream; others, such as M. tuberculosis, are macrophage-tropic. Plasmodium replication in hepatocytes and erythrocytes is highly iron-dependent (many other pathogen types also target hepatocytes because they are energy- and nutrient-rich). Fluctuations in hepcidin – directly consequent to infection as part of the innate immune response, or due to the underlying iron status of the infected individual, or caused by experimental or therapeutic intervention – alter iron availability to the infectious organism, with likely downstream effects on pathogenesis. Iron is also required for host immune mechanisms to generate microbicidal effectors (reactive oxygen species, nitric oxide) and for antigen-specific lymphocyte proliferation. Image credits: Wikimedia, CDC, NIH; bacteria images copyright Dennis Kunkel Microscopy Inc. Reproduced with permission from Drakesmith and Prentice [23] .
Competing Control of Iron by Iron Needs and Infection/Inflammation
Hepcidin represents an exquisite example of evolution in response to the 2 opposing aspects of iron in biology; its obligate need for many physiological processes versus the threat it poses by encouraging pathogen growth. Existing data suggest that hepcidin balances the need for iron against the threat of infection [23]. Figure 3 (reproduced from Wallace [11] ) and its accompanying legend gives a simplified summary of the mechanisms by which hepcidin achieves this balancing act. The figure summarizes current knowledge and numerous further details are sure to emerge.
In brief, the iron-responsive modulation derives signals from hepatic iron and transferrin saturation and possibly oxygen saturation through HIF1. The recently described bone marrow-derived hormone, erythroferrone, produced by young erythroblasts also downregulates hepcidin expression in response to active erythropoiesis
[24]. Conversely, inflammation, mostly mediated through IL-6 but with alternative stimulation by IL-22 and type 1 interferon, upregulates hepcidin and thus blocks iron absorption and recycling [25] . Figure 4 illustrates the impressive speed and extent by which hepcidin can elicit an immunoprotective innate response [26] .
Figure 5, reproduced from Drakesmith and Prentice [23], illustrates how differential levels of hepcidin expression change the iron concentration in various body compartments with likely consequences for the susceptibility to infection. Hepcidin-induced hypoferremia in circulating plasma protects against the acute threat of bacterial and yeast sepsis. Prolonged upregulation of hepcidin leads to the anemia of inflammation or of chronic infection [27], which in turn protects against the blood stage of malarial infection [28] . Similarly, hypoferremia in hepatocytes protects against the hepatic stages of malaria infection [29] . Intriguingly, though as yet unproven, we propose that iron lock-down in macrophages may be the reason that intracellular pathogens have selected macrophage phagosomes as their niche of choice [23].
New Insights into the Causes of Iron Deficiency in Low-Income Countries
Prior to this century, almost all research into the etiology of iron deficiency anemia concentrated on dietary factors and was based around the premise, as mentioned above, that humans are inherently badly designed to absorb iron especially from cereal-based diets. The discovery of the hepcidin-ferroportin axis effectively turns this assumption on its head and requires a thorough re-examination of the key etiological factors driving deficiency. Key to this conclusion is the finding that most known gene defects affecting iron status lead to iron overload, not deficiency, even in populations on a low iron intake [11]. The one exception relates to defects in the TMPRSS6 gene which normally suppresses hepcidin gene expression. Rare defects in this gene result in hep-cidin overexpression and so-called iron-refractory iron deficiency anemia (IRIDA) [30] . These insights reveal that instead of struggling to absorb sufficient iron, the normal physiological state in humans is one of tonic suppression of iron absorption in order to maintain body iron homeostasis in the absence of any pathways for secretion of an excess.
We have extensively studied populations with high levels of anemia (pregnant women and young children in rural areas of Gambia and Kenya) and have been forced to re-evaluate many of our preconceived notions. First, in cross-sectional studies in both populations and using structural equation modelling we showed that hepcidin levels were, as predicted on the basis of the known biology of hepcidin, predicted by a combination of iron status, inflammation, and infection [31, 32].
We then used receiver-operator characteristic analysis to determine hepcidin cutoffs that best define iron deficiency anemia and the anemia of inflammation [33, 34] . In children the defined threshold (5.5 ng/mL, using the Bachem ELISA) also distinguished iron absorbers from nonabsorbers [33] . With support from the Bill & Melinda Gates Foundation, we have recently completed 2 randomized controlled trials in Gambian pregnant women and children under the umbrella of the HIGH Consortium (Hepcidin and Iron in Global Health) [35, 36] . These trials are testing whether it would be possible to develop a point-of-care diagnostic for hepcidin-guided iron administration on the principle that a hepcidin level below 5.5 ng/mL in children (or 2.5 ng/mL in pregnant women [34]) would signal that the subject was “safe and ready to receive iron.” These trials will shorty report full results.
Using longitudinal hepcidin measurements (weekly for 12 weeks) in over 400 participants from the children’s trial [35] we have shown that each child maintains a remarkably consistent level of hepcidin over the 3 months of study (albeit gradually rising in response to the iron supplementation). Fifty percent of the children consistently maintain their hepcidin above the 5.5 ng/mL, indicating that they are blocking iron absorption (Prentice et al., manuscript in preparation). Unsurprisingly, these high hepcidin levels are associated with raised CRP and AGP (markers of inflammation), but remarkably we found that hepcidin was upregulated even at very low levels of inflammation. This has important implications for the prevention and treatment of iron deficiency and its associated anemia as we discuss in our conclusions below.
Prospects and Possible Applications for Hepcidin Agonists and Antagonists
Pharmaceutical companies are investing considerable resources in the potential therapeutic applications that could be derived from manipulating hepcidin [37] . For instance, iron overload could potentially be modulated by artificially enhancing hepcidin. To this end, Ganz and his team have synthesized a series of mini-hepcidins; short peptides that maintain hepcidin action, are re-engineered to be stable, and could potentially be orally active [38, 39] . In the global health context mini-hepcidin might be able to prevent the iron overload that occurs even in untrans-fused beta-thalassemia (due to the hepcidin-suppressing action of excess erythroferrone caused by the ineffective erythropoiesis that generates excess erythroblasts) [40] . Conversely, there is great interest in the possibility that hepcidin antagonists could be used to prevent or treat the anemia of chronic disease and anemias associated with treatments such as cancer chemotherapy [37] . Clinical trials are underway for both hepcidin agonists and antagonists and it is likely that these will become commonly used therapeutic agents in the mid-term future.
Conclusions: Implications of the New Molecular Insights
It goes without saying that the design and implementation of preventative or therapeutic interventions to combat disorders of iron metabolism (most commonly iron deficiency anemia in a global context) are greatly aided by a clearer understanding of the precise mechanisms regulating iron metabolism. As described above, the most important implication of these discoveries is the realization that humans have evolved to expend at least as much physiological effort in excluding dietary iron as in acquiring it. This has been driven by the hazardous nature of iron; both in causing oxidative damage and in promoting the growth of microorganisms. There is clear evidence that a moderate degree of iron deficiency anemia is highly protective against malaria [28] and likely protective against a range of bacterial and fungal pathogens [23] and possibly viruses [41] . This has resulted in the evolution of exquisite systems for chaperoning iron and regulating its intake and organ distribution. Against this background, our former attempts to counteract iron deficiency by using very high nonphysiological doses of highly absorbable iron given without food seem clumsy to say the least and have almost certainly caused a great deal of iatrogenic harm. An analogy would be that we have been trying to use a sledgehammer to break down a closed door rather than learning how to pick the lock. Our new understanding that even low level inflammation blocks iron absorption via the hepcidin/ferroportin axis throws the spotlight on the need to eliminate infections and the consequent inflammation. This will require so-called nutrition-sensitive interventions around improvements in hygiene and infection control and it is quite possible that such interventions could be more important in poor populations than nutrition-specific interventions around iron.
Disclosure Statement
The author is a member of the Nestlé Nutrition Institute (NNI) Board and receives an honorarium as faculty for the annual NNI Training Course in Pediatric Nutrition. He has also consulted for Danone, Wyeth, and Vifor.
Funding Sources
The author is supported by MCA760-5QX00 to the MRC International Nutrition Group by the UK Medical Research Council (MRC) and the UK Department for International Development (DFID) under the MRC/DFID Concordat agreement.
References
-
Schade AL: Raw hen egg white and the role of iron in growth inhibition of Shigella dysenteriae, Staphylococcus aureus, Escherischia coli and Saccharomyces cerevisiae. Science 1944; 100: 340–341.
-
Schade AL, Caroline L: An iron-binding component in human blood plasma. Science 1946; 104: 340–341.
-
McCance RA, Widdowson EM: Absorption and excretion of iron. Lancet 1937; 230: 680– 684.
-
Widdowson EM, McCance RA: Iron exchanges of adults on white and brown bread diets. Lancet 1942; 239: 588–591.
-
Cavell PA, Widdowson EM: Intakes and excretion of iron, copper and zinc in the neonatal period. Arch Dis Child 1964; 39: 496– 501.
-
Turnland JR: The use of stable isotopes in mineral nutrition research. J Nutr 1989; 119: 7–14.
-
Kastenmayer P, Davidsson L, Galan P, Cher-ouvrier F, Hercberg S, Hurrell RF: A double stable isotope technique for measuring iron absorption in infants. Br J Nutr 1994; 71: 411– 424.
-
Hunt JR: Iron. Encyclopedia of Human Nutrition, ed 3. Academic Press, 2005, vol 3, pp 82–89.
-
Prentice AM: Iron metabolism, malaria, and other infections: what is all the fuss about? J Nutr 2008; 138: 2537–2541.
-
Gunshin H, Mackenzie B, Berger UV, Gun-shin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA: Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997; 388: 482– 488.
-
Wallace DF: The regulation of iron absorption and homeostasis. Clin Biochem Rev 2016; 37: 51–62.
-
McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, et al: An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001; 291: 1755–1759.
-
Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC: The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1: 191– 200.
-
Osaki S, Johnson DA, Frieden E: The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J Biol Chem 1966; 241: 2746–2751.
-
Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, et al: Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21: 195–199.
-
Hentze MW, Muckenthaler MU, Andrews NC: Balancing acts: molecular control of mammalian iron metabolism. Cell 2004; 117: 285–297.
-
Ganz T: Systemic iron homeostasis. Physiol Rev 2013; 93: 1721–1741.
-
Krause A, Neitz S, Mägert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K: LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett 2000; 480: 147–150.
-
Park CH, Valore EV, Waring AJ, Ganz T: Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001; 276: 7806–7810.
-
Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loréal O: A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 2001; 276: 7811–7819.
-
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan
J:Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004; 306: 2090–2093.
-
Prentice AM, Doherty CP, Abrams SA, Cox SE, Atkinson SH, Verhoef H, Armitage AE, Drakesmith H: Hepcidin is the major predictor of erythrocyte iron incorporation in anemic African children. Blood 2012; 119: 1922– 1928.
-
Drakesmith H, Prentice AM: Hepcidin and the iron-infection axis. Science 2012; 338: 768–72.
-
Kautz L, Jung G, Valore EV, Rivella S, Ne-meth E, Ganz T: Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 2014; 46: 678–684.
-
Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T: IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113: 1271–1276.
-
Rivera S, Nemeth E, Gabayan V, Lopez MA, Farshidi D, Ganz T: Synthetic hepcidin causes rapid dose-dependent hypoferremia and is concentrated in ferroportin-containing organs. Blood 2005; 106: 2196–2199.
-
Ganz T, Nemeth E: Hepcidin and iron homeostasis. Biochim Biophys Acta 2012; 1823: 1434–1443.
-
Goheen MM, Wegmüller R, Bah A, Darboe B, Danso E, Affara M, Gardner D, Patel JC, Prentice AM, Cerami C: Anemia offers stronger protection than sickle cell trait against the erythrocytic stage of falciparum malaria and this protection is reversed by iron supplementation. EBioMedicine 2016; 14: 123–130.
-
Portugal S, Carret C, Recker M, Armitage AE, Gonçalves LA, Epiphanio S, Sullivan D, Roy C, Newbold CI, Drakesmith H, Mota MM: Host-mediated regulation of superinfection in malaria. Nat Med 2011; 17: 732–737.
-
Finberg KE, Heeney MM, Campagna DR, Aydinok Y, Pearson HA, Hartman KR, et al: Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet 2008; 40: 569–571.
-
Atkinson SH, Armitage AE, Khandwala S, Mwangi TW, Uyoga S, Bejon PA, Williams TN, Prentice AM, Drakesmith H: Combinatorial effects of malaria season, iron deficiency, and inflammation determine plasma hepcidin concentration in African children. Blood 2014; 123: 3221–3229.
-
Atkinson SH, Uyoga SM, Armitage AE, Khandwala S, Mugyenyi CK, Bejon P, Marsh K, Beeson JG, Prentice AM, Drakesmith H, Williams TN: Malaria and age variably but critically control hepcidin throughout childhood in Kenya. EBioMedicine 2015; 2: 1478– 1486.
-
Pasricha SR, Atkinson SH, Armitage AE, Khandwala S, Veenemans J, Cox SE, Eddow-es LA, Hayes T, Doherty CP, Demir AY, Tijhaar E, Verhoef H, Prentice AM, Drake-smith H: Expression of the iron hormone hepcidin distinguishes different types of anemia in African children. Sci Transl Med 2014; 6: 235re3.
-
Bah A, Pasricha SR, Jallow MW, Sise EA, We-gmuller R, Armitage AE, Drakesmith H, Moore SE, Prentice AM: Serum hepcidin concentrations decline during pregnancy and may identify iron deficiency: analysis of a longitudinal pregnancy cohort in The Gambia. J Nutr 2017; 147: 1131–1137.
-
Wegmüller R, Bah A, Kendall L, Goheen MM, Mulwa S, Cerami C, Moretti D, Prentice AM: Efficacy and safety of hepcidin-based screen-and-treat approaches using two different doses versus a standard universal approach of iron supplementation in young children in rural Gambia: a double-blind randomised controlled trial. BMC Pediatr 2016; 16: 149.
-
Bah A, Wegmuller R, Cerami C, Kendall L, Pasricha SR, Moore SE, Prentice AM: A double blind randomised controlled trial comparing standard dose of iron supplementation for pregnant women with two screen-and-treat approaches using hepcidin as a biomarker for ready and safe to receive iron. BMC Pregnancy Childbirth 2016; 16: 157.
-
Sebastiani G, Wilkinson N, Pantopoulos K: Pharmacological targeting of the hepcidin/ ferroportin axis. Front Pharmacol 2016; 7: 160.
-
Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, Sharma S, Waring A, Ganz T, Nemeth E: Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest 2011; 121: 4880–4888.
-
Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, Ganz T: Minihepci-dins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 2012; 120: 3829–3836.
-
Casu C, Oikonomidou PR, Chen H, Nandi V, Ginzburg Y, Prasad P, Fleming RE, Shah YM, Valore EV, Nemeth E, Ganz T, MacDonald B, Rivella S: Minihepcidin peptides as disease modifiers in mice affected by ß-thalassemia and polycythemia vera. Blood 2016; 128: 265– 276.
-
Drakesmith H, Prentice A: Viral infection and iron metabolism. Nat Rev Microbiol 2008; 6: 541–552.