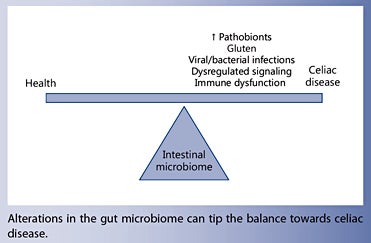
Intestinal dysbiosis is a hallmark of several immune disorders, including celiac disease. Increased levels of pathobionts may activate the proinflammatory pathways that trigger a breakdown in gluten tolerance and promote disease onset. This, in turn, further drives the gut microbial community towards a state of dysbiosis, resulting in a vicious circle that disrupts the host-microbiota homeostatic balance. The genes that predispose an individual towards celiac disease modulate the infant’s gut colonization process, highlighting the importance of the gut microbiome in tipping the balance towards health or disease.
Current knowledge
The gut microbiome plays a key role in the host’s defense mechanisms against pathogens and is also involved in the maturation, maintenance and function of the mucosal immune system. The microbiome is a highly complex entity that exhibits many redundant functions and is capable of evolving in response to genetic and environmental factors. Thus, although a large body of literature exists on the features of a healthy microbiome, it is difficult to pinpoint the microbial signature of celiac disease. Further research is needed to understand how intestinal bacteria interact within the host environment to promote celiac disease.
Practical implications
Although gluten is the main environmental trigger, the timing of disease onset following gluten exposure varies greatly between individuals, suggesting that other factors are involved. The pathogenic mechanisms that contribute towards celiac disease may be triggered by viral or bacterial infections that could amplify the response to gluten in predisposed individuals. In addition, individuals with celiac disease have perturbations in the gut microbial signature, such as abnormal levels of Bacteroides spp., Bifidobacterium spp. and Staphylococcus spp. Not surprisingly, celiac disease patients also show alteration in the levels of metabolites (i.e. short-chain fatty acids) and molecules (α-defensins, TLR2, TLR4) involved in host-microbial interactions.
Recommended reading
Hooper LV, Littman DR, Macpherson AJ: Interactions between the microbiota and the immune system. Science 2012;8;336:1268–1273.
Celiac disease (CD) is associated with intestinal dysbiosis characterized by increases in the abundance of pathobionts and their virulence features.
Infections and expansion of intestinal pathobionts could theoretically activate inflammatory pathways that precipitate the breakdown of gluten tolerance or aggravate CD pathogenesis.
CD susceptibility genes influence the infant’s gut colonization process, suggesting that predisposed individuals may have a compromised ability to acquire a protective microbiota.
Celiac disease (CD) is a frequent chronic inflammatory enteropathy caused by gluten in genetically predisposed individuals that carry disease susceptibility genes (HLADQ2/ 8). These genes are present in about 30–40% of the general population, but only a small percentage of carriers develops CD. Gluten is the key environmental trigger of CD, but its intake does not fully explain disease onset; indeed, an increased number of cases experience gluten intolerance in late adulthood after many years of gluten exposure. Consequently, additional environmental factors seem to be involved in CD. Epidemiological studies indicate that common perinatal and early postnatal factors influence both CD risk and intestinal microbiota structure. Prospective studies in healthy infants at risk of developing CD also reveal that the HLA-DQ genotype, in conjunction with other environmental factors, influences the microbiota composition. Furthermore, CD patients have imbalances in the intestinal microbiota (dysbiosis), which are not fully normalized despite their adherence to a gluten-free diet. Therefore, it is hypothesized that the disease can promote dysbiosis that aggravates CD pathogenesis, and dysbiosis, in turn, can initiate and sustain inflammation through the expansion of proinflammatory pathobionts and decline of anti-inflammatory mutualistic bacteria. Studies in experimental models are also contributing to understand the role of intestinal bacteria and its interactions with a predisposed genotype in promoting CD. Advances in this area could aid in the development of microbiome-informed intervention strategies that optimize the partnership between the gut microbiota and host immunity for improving CD management.
Introduction
The human intestinal tract harbors a complex microbiota that develops a symbiotic mutualistic relationship with its host that benefits both organisms in physiological conditions. The intestinal microbiota is integrated by approximately 5–7 main bacterial phyla of the 52 currently recognized on Earth. In adults, Firmicutes and Bacteroidetes are generally the dominant phyla (~90% of the total population), followed by members of Proteobacteria and Actinobacteria, which are by far less abundant (<1– 5%) [1]. The phylum Bacteroidetes contains the wellknown genera Bacteroides and Prevotella . Firmicutes constitute the largest bacterial phylum, which contains more than 200 genera, including clostridial clusters. Proteobacteria are facultative anaerobic bacteria that include the well-known Enterobacteriaceae family and may represent only ~0.1% of the total population. The phylum Actinobacteria includes the genus Bifidobacterium and its abundance largely varies with age. The number of bacterial species is estimated to be larger than 1,000 and the number of genes (collectively known as ‘microbiome’) is more than 150-fold greater than that of the human genome [2]. This vast repertoire of microbiome genes provides the host with complementary functional resources, such as pathways for nutrient utilization (e.g. complex polysaccharides) and vitamin production as well as molecules instructing intestinal morphological and immune maturation [2, 3]. Consequently, it is thought that the intestinal microbiota composition and function play an important role in the balance between the host’s health and disease.
The colonization process of the newborn intestine primarily occurs after birth and represents a major exposure to environmental antigens that influences the development of mucosal and systemic immunity. The exposure to a highly dense microbial population within the gut also represents a major challenge to the host, which needs to develop complex control mechanisms to eliminate pathogens and tolerate symbionts. It is hypothesized that the dysregulation of these reciprocal interactions increases the risk of developing immune-mediated disorders.
The colonization of the infant’s gut is generally characterized by the initial dominance of facultative anaerobes (mainly Proteobacteria species) that make the habitat suitable for colonization by strict anaerobes by consuming oxygen, altering the pH and producing carbon dioxide and nutrients [4]. Within the first week after birth, those are rapidly replaced by strict anaerobes of which Actinobacteria become dominant when infants are being milk-fed and especially in those being breastfed due to the stimulation of the growth of Bifidobacterium spp. by breast milk oligosaccharides [4]. Cessation of milk feeding, which implies the incorporation of solid food, drives full maturation of the microbiota towards a complex adult-like one, dominated by Firmicutes and Bacteroidetes [5]. This process depends, however, on multiple factors including genetic and environmental variables (type of birth, intake of antibiotics, milk-feeding practices) [4, 5]. Early exposure to those environmental factors seems to be an important determinant of health, since the microbiota is being established when the immune system of the neonate is immature. Through this process, tolerance (hyporesponsiveness or mutualistic response) to innocuous antigens from the diet and the microbiota is developed, facilitating the establishment of a protective microbiota, while ensuring elimination of harmful antigens and pathogens by developing an active proinflammatory response [6].
Experimental models have demonstrated that gut microbiota influence the development and function of different host defense mechanisms of innate and adaptive immunity [3, 7]. Gut microbiota influences the role of intestinal epithelial cells as first line of defense by different mechanisms. These include regulation of epithelial cell proliferation and expression of tight junction proteins that held those cells together controlling paracellular permeability. These constitute a physical barrier that separates the intestinal lumen from the underlying tissue, where the gut-associated lymphoid tissue is located, avoiding its activation that may cause chronic inflammation. Gut microbiota also influences mucin gene expression (e.g. MUC-2 and MUC-3 genes) by goblet cells and their glycosylation pattern and secretion of antimicrobial peptides (defensins, angiogenins, Reg3γ, etc.) by intestinal cells, which contribute to regulating bacterial adhesion and residence in the gut. Furthermore, specific components of the gut microbiota also affect the expression and activation of pattern recognition receptors, such as Toll-like receptors (TLRs), which are expressed by epithelial cells and innate immune cells [e.g. dendritic cells (DCs)] and play an important role in maintaining a host-microbiota homeostatic cross talk [8]. Activation of TLRs may lead to the synthesis of various proinflammatory cytokines and chemokines via transcriptional regulation and constitutes one of the mechanisms by which the host tolerates innocuous microbes while simultaneously preventing their dissemination and infections. TLR-mediated innate immune responses are also a prerequisite for the generation of adaptive immune responses [8].
Experimental models have also proved that the gut microbiota impacts on adaptive immunity. Thus, the absence of microbiota leads to a lower proportion of the lamina propria T cells, IgA-producing B cells and intraepithelial T cells and serum immunoglobulin levels, while other cell subpopulations may not be affected or even increase in germ-free conditions [3, 9] . In recent years, specific commensal bacteria have been shown to influence the makeup of lamina propria Tlymphocyte subsets that have distinct effector functions, either potentially proinflammatory (including Th1, Th17 and diverse innate lymphoid cells with cytokine effector features) or anti-inflammatory regulatory T cells (Tregs) [10] . In particular, the induction of colonic Foxp3+ Tregs is mediated through the activation of anti-inflammatory cytokine production by epithelial and innate immune cells (DCs). Then, the gut microbiota structure could play a major role in the regulation of diverse immune homeostatic host control mechanisms and, therefore, in the development of immunemediated diseases.
In humans, prospective studies have also suggested that the disturbance of the colonization process is related to an increased risk of immune-mediated diseases [6]. Associations have been established between the intestinal colonizing process in infants and the production of salivary secretory IgA, numbers of circulating IgA and IgM antibody-producing cells and the expression of innate immune receptors (e.g. TLR) in peripheral blood monocytes, which suggest a possible mechanism by which the microbiota confers immune maturation signals and protection in humans at early stages of life [6].
It has also been proposed that immune dysfunction, characteristic of overt autoimmune and other chronic inflammatory disorders, drives the microbial community to a state of dysbiosis that usually implies both alterations in microbiota structure and loss of stability. Indeed, a number of studies reveal that intestinal dysbiosis and inflammation are associated [11–13], supporting a role of alterations in the host-microbiota cross talk in immunemediated disorders. From a mechanistic point of view, a disturbed microbiota could play a role in autoimmunity through a variety of potential mechanisms, including molecular mimicry, based on cross-reactivity of effector lymphocytes to microbial and host or dietary peptides or proteins; bystander activation of adaptive autoimmunity by antigen-presenting cells activated by microbial stimuli and expressing self-antigens, and by loss of innate control of the host-commensal symbiotic relationship leading to interference in the activation of innate immune mechanisms [13].
A genetic predisposition is present in about 30–40% of the general population, and only a small percentage of carriers (2–5%) develops CD
The prototypical gastrointestinal autoimmune disorder is celiac disease (CD). This is a frequent chronic inflammatory enteropathy caused by gluten in genetically predisposed people that carry disease susceptibility genes (HLA-DQ2/8). It is now accepted that both innate and adaptive mechanisms, where antigen-presenting T-cell interaction is central, are necessary for developing the disorder. However, a genetic predisposition is present in about 30–40% of the general population, and only a small percentage of carriers (2–5%) develops CD. Gluten is the main environmental trigger of CD, but its intake does not fully explain the disease onset. In fact, while some subjects develop the disease after the introduction of gluten into the diet, others do so in late adulthood after having been exposed to gluten for many years, suggesting that other disease-promoting factors are involved. Emerging evidence suggests that the triad that may better explain the likelihood of developing CD and the disease spectrum must be integrated by signals and functions encoded by the so-called second human genome (microbiome) and other environmental factors that, in conjunction, could regulate the pathogenic pathways activated by gluten in predisposed individuals [14] . In this review, we discuss the parallelism between the CD pathogenic mechanisms activated by gluten and potentially by intestinal pathogenic microbes, the human microbiome features of CD and the current evidence of a pathogenic role of intestinal dysbiosis inferred from CD experimental study models.
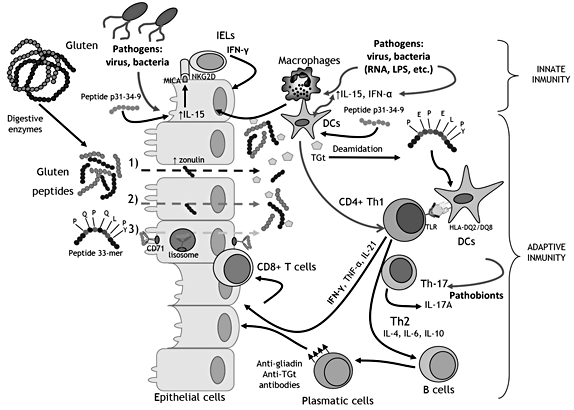
Fig. 1. Schematic representation of the proposed model for CD pathogenesis. Gray arrows indicate how pathogens (virus and bacteria) and pathobionts (potentially pathogenic intestinal bacteria) could theoretically cooperate in driving CD pathogenesis. TGt = Tissue transglutaminase; IEL = intraepithelial lymphocytes; NKG2D = natural killer group 2D receptor; MICA = MHC class I chain-related protein A; CD71 = cluster of differentiation 71 or transferrin receptor protein 1; DCs = dendritic cells.
Gluten and Microbes: How Both Could Cooperate in Driving CD Pathogenesis
CD is a T-cell-mediated disorder triggered by a group of proteins generally termed gluten contained in wheat, barley and rye. Genetic predisposition is necessary for the development of CD and occurs in individuals carrying the HLA-DQ2 (HLA-DQ2.5 and HLA-DQ2.2) and HLADQ8 haplotypes. After ingestion, gluten proteins are partially hydrolyzed during digestion due to their high content of glutamine and proline residues. Then, peptides are accumulated in the intestinal lumen and are responsible for the activation of a deregulated immune response characteristic of CD. The proposed model of CD pathogenesis and the possible contribution of infectious agents or pathobionts are schematized in figure 1.
Some gluten-derived peptides have been proposed to activate innate immunity in the intestinal epithelium without inducing T-cell-specific responses. For example, α-gliadin 31–43 peptide induces the production of IL-15 by innate cells (epithelial cells, macrophages and DCs) that, in turn, activate cytotoxic cells [i.e. intraepithelial lymphocytes (IELs) CD8+] independently of the T-cell receptor specificity. In the epithelium, IL-15 upregulates the non- MHC class I natural killer receptor NKG2D and CD94 on IELs and licenses these cells to kill epithelial cells expressing the stress-inducible ligand MICA; IL-15 also makes IELs resistant to the inhibitory effects of counter-regulatory mechanisms mediated by Tregs [15, 16] . However, it is generally thought that IELs do not mediate tissue damage through gluten recognition since evidence of the existence of gluten-peptide epitopes recognized by CD8+ T cells is limited; therefore, other possible ligands of IELs have been proposed, like self-antigens and virus acting as CD-promoting factors [15] . Nevertheless, this issue remains controversial, as a clear pathogenic pathway and receptor for these innate peptides have not been described in vivo.
Additional innate immunity cytokines are overproduced in CD mucosa and supposed to amplify the inflammation and tissue-damaging response. These include IL-1 cytokine family members (IL-1β and IL-18), TNF-α and IL-6, some of which seem to be involved particularly in refractory CD cases unresponsive to a gluten-free diet (GFD) [17, 18]. The increased secretion of these proinflammatory cytokines may involve the activation of innate immune signaling pathways by gluten peptides (e.g. peptide P31–43) that involve the myeloid differentiation factor 88 (MyD88), a key adapter molecule of the TLR/IL- 1R pathways, but without depending neither on TLR2 nor TLR4 [19]. Other studies have also suggested that signaling pathways downstream TLRs, including MyD88/TRIF/ MAPK/NF-κB, may be involved in the responses to some innate gluten peptides [17]. Interestingly, these pathways also respond to bacterial motifs that are recognized by TLRs expressed in epithelial and innate immune cells [8]. An α-amylase/trypsin inhibitor found in wheat was also shown to interact with TLR4 and stimulate macrophages and DCs to produce some of these inflammatory cytokines, suggesting that non-gluten components of wheat may also activate innate immune responses similarly to microbes [20]. Accumulation of innate lymphoid cells located in the lamina propria of duodenal biopsies from CD patients also seems to contribute to the inflammatory process and intestinal damage by rapidly producing TNF-α, in addition to IFN-γ [21] . Moreover, this ex vivo study with duodenal biopsies showed that TLR-3 mediated the production of TNF-α in response to the syn thetic analog of double- stranded RNA (poly I:C), simulating a viral infection, suggesting that microbial agents can also contribute to the inflammatory CD response [21] . To mimic the virally induced activation of TLR3 signaling, other studies evaluated the effects of mucosal administration of poly I:C in mouse models. This also caused enteropathy and gut dysfunction in gliadin-sensitive NOD-DQ8 mice, which was exacerbated by the subsequent oral delivery of gluten, pointing in the same direction [22].
Gluten-derived peptides need to gain access to the lamina propria to stimulate the full immune response characteristic of CD. Although it has been suggested that peptides cross the epithelium by transcellular transport [23] and probably by paracellular pathways [24], the exact mechanism remains unclear. In the lamina propria, gluten-derived peptides are recognized by HLA-DQ molecules expressed by antigen-presenting cells (DCs), thereby activating a gluten-specific inflammatory CD4 Th1 response with production of proinflammatory cytokines, mainly IFN-γ. This inflammatory milieu also activates cytotoxic CD8 IELs (CD8+ TCRαβ+ and TCRγδ+ T cells) in the lamina propria, and contributes to expand the IEL population independently of antigen presentation via MHC, and the synthesis and release of metalloproteases and keratinocyte growth factor by stromal cells that lead to crypt hyperplasia and enteropathy. IL-21, a cytokine produced by gluten- specific CD4 T cells, has recently been reported to be increased in CD mucosa. This cytokine could play a role in CD pathogenesis by enhancing IFN-γ production in CD4+ T cells, increasing the number of intraepithelial CD8+ T cells, contributing to CD-associated autoantibody production and increasing the resistance of inflammatory T cells to Tregs [25]. IL-21 has been shown to increase TLR4 expression in in vitro cell cultures, and, theoretically, this could also promote TLR4 signaling by intestinal bacteria in CD, since tissue damage increases contact with microbeassociated molecular patterns [26]. In biopsies from pediatric CD patients, stimulation with TLR3 ligands, simulating viral stimulation, during polyclonal T-cell activation significantly increased IL-21 secretion, suggesting a potential role of viral infections [25]. IL-17A overproduction, which is a cytokine synthesized by non-gluten-reactive CD4+ T cells and CD8+ T cells, has also been shown to be elevated at the late stage of acute CD when villous atrophy has developed, but not in potential CD or in CD patients on a GFD, suggesting that it is not primarily involved in CD [25]. This cytokine is also upregulated in refractory CD, where symptoms/signs of malabsorption and villous atrophy persist or recur after long-term adherence of a GFD, while this is not the case with Th1-type cytokines [18]. Th17 cell differentiation from naïve T cells requires the coordinated action of cytokines as those involved in the CD inflammatory process (e.g. IL-21), which could be responsible for the upregulation of Th17-type cytokines such as IL-17A. Moreover, Th17 cells have critical functions in host defense against pathogens, particularly those encountered at mucosal surfaces and specific intestinal bacteria (e.g. segmented filamentous bacteria) are known to act as strong promoters of differentiation of naïve CD4+ T cells into Th17 cells and IL-17A production in experimental models [27]. Therefore, theoretically, intestinal bacteria could also contribute to the activation of IL- 17A response in the intestinal mucosa of active CD patients and in subjects unresponsive to the GFD [28].
IL-15 is a cytokine that has been proposed to be critical not only in the link between the innate and adaptive immune response in CD. In fact, IL-15 is upregulated in both the epithelium and the lamina propria in a proportion of patients with active CD, where they can polarize DCs towards a proinflammatory phenotype and activate inflammatory cytokine release (IFN-γ, IL-21) by lamina propria CD4+ and CD8+ lymphocytes [16, 29]. IL-15 can also be induced in DCs via the activation of TLR4 with LPS from Gram-negative bacteria and via the MyD88 pathway in epithelial cells [16], suggesting a potential link with intestinal bacterial products (LPS) or microbes which could also be sampled by DCs from the intestinal lumen.
IFN-α can also prime DCs and their interactions with T cells in the lamina propria, leading to either an inflammatory response by inducing the expansion of glutenspecific CD4+ T cells and activating cytotoxic lymphocytes (CD8+ cells) [30]. Existing evidence also suggests a role of IFN-α in the DC-mediated activation of IFN-γ production by T cells in the lamina propria. In fact, IFN-α is known to be highly expressed in the small bowel of CD patients; blocking IFN-α inhibits IFN-γ mRNA expression in organ cultures of CD biopsies, and type-I IFN activates DCs towards an inflammatory phenotype [16]. Rotavirus and pathogenic bacteria are known to induce type-I IFNs. Indeed, signaling through TLRs upon interaction with LPS from Gram-negative bacteria or with viral RNA can activate interferon regulatory factors and lead to the production of type-I IFNs, such as IFN-α [8, 31].
Cytokines and pathways contributing to CD pathogenesis can also be induced by viral and bacterial infections or intestinal pathobionts that could theoretically amplify the response to gluten in predisposed individuals
In summary, cytokines and pathways contributing to CD pathogenesis can also be induced by viral and bacterial infections or intestinal pathobionts that could theoretically amplify the response to gluten in predisposed individuals. Some epidemiological studies support this idea, as incidence of rotaviral infections in childhood has been shown to increase CD risk and other gastrointestinal infections have been reported at the time of CD diagnosis [6] . Nevertheless, these theories are rather speculative, and more direct evidence is needed.
Microbiome Features of CD
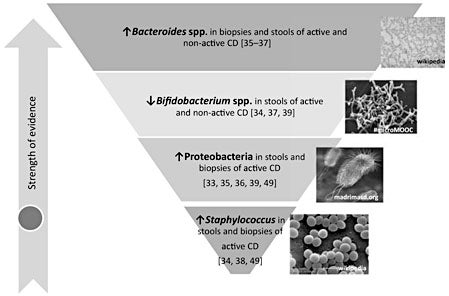
Studies on the microbiome structure of subjects with active CD (newly diagnosed patients under a GFD) and non-active CD (patients under a GFD) generally support that this chronic enteropathy is associated with intestinal dysbiosis that, in turn, could aggravate and even promote the disease onset. Some microbiota perturbations have been consistently found in subjects with active and nonactive CD, suggesting a role in promoting disease development. Other perturbations seem to be normalized after long-term adherence to a GFD, suggesting that there must be a secondary consequence of the inflammatory process associated with the disease that could aggravate this disorder. The fact that the perturbations in gut microbiota are more pronounced in the active phase of CD demonstrates the activation of a loop of pathogenic events that alters the host-microbe cross talk and, presumably, may influence CD manifestation and evolution. In fact, some studies have identified microbiota alterations particularly in subjects with persistent symptoms despite adherence to a long-term GFD [32] or associated with gastrointestinal symptoms but not with dermatitis herpetiformis [33]. The expansion of specific bacterial groups with potential pathogenic features may represent an active component of the pathogenic process rather than a mere passive consequence of the disease. Figure 2 schematizes the strength of existing evidence of an association between CD and intestinal dysbiosis and the potential taxonomic groups that may constitute biomarkers of CD. The evidence was graded according to the population groups compared (CD subjects treated or not with a GFD), the number of studies and consistency across at least two studies and sampling sites (intestinal biopsies or stools).
Fig. 2. Presumptive microbiome-based biomarkers of CD. These are supported by at least two independent studies.
Studies published to date demonstrate that increases in the abundance of Bacteroides spp. and decreases in Bifidobacterium spp. in small intestinal biopsies and/or stool samples characterize the microbiota of CD, regardless of the state of the disease (active or non-active). In particular, our studies using quantitative PCR found that Bacteroides spp. were more abundant in feces and biopsies CD patients with active and non-active disease than in controls [34]. The differential bacterial groups detected in CD biopsies and feces correlated, indicating that the fecal microbiota partly reflects that of the small intestine and may have a diagnostic value [34, 35]. Duodenal biopsy analysis by temporal temperature gradient gel electrophoresis (TGGE) also demonstrated a higher prevalence of Bacteroides vulgatus in children with active and non-active CD [36]. An independent study based on the cultural analysis of stool samples also found increased Bacteroides numbers in untreated and treated CD patients compared to controls [37]. Other studies only reported increases in Bacteroides spp. in untreated CD patients compared to controls because only these two groups of subjects were compared [38, 39]. However, in other cases, differences were reported to be statistically significant only between the biopsy samples of patients with active disease and controls, likely because the differences were ameliorated after adherence to the GFD and the analytic techniques used have a higher detection limit (FISH), which reduce the ability to detect the smaller differences, although the same trends were detected [40, 41]. Comparisons of treated CD patients and healthy subjects also demonstrated increases in presumptive Bacteroides numbers in the stool of the patients, confirming previous findings, but untreated CD cases were not included [39]. We also carried out a deeper characterization of the CD microbiota by isolating bacterial strains and analyzing their pathogenic features. Specifically, we found that the abundance of the species Bacteroides fragilis coding for metalloproteases was increased in both untreated and treated CD patients, and thus could presumably play a pathogenic role in CD [42]. In fact, Bacteroides fragilis strains coding for virulence factors (e.g. metalloproteases) are frequently involved in opportunistic infections and were demonstrated to aggravate colitis in experimental animal models [42].
In contrast, a study that analyzed the duodenal microbiota of CD patients suffering from persistent symptoms despite a long-term GFD by 16S rRNA gene pyrosequencing showed that the patients had a lower abundance of Bacteroidetes [32]. Nevertheless, no other studies have been conducted in this category of subjects to draw definitive conclusions. Other studies did not report differences in Bacteroides , but they used semi-quantitative and lower-resolution techniques that only detect dominant bacteria [denaturing gradient gel electrophoresis (DGGE)] [33] or include a very low number ( ≤ 10) of cases [33, 43–45]. Some of those studies detected differences in a related bacterial genera Prevotella spp., which belongs to the same phylum Bacteroidetes, but the results are inconsistent and contradictory across studies [43–45]. A Swedish study of samples collected in the CD epidemic between 1985 and 1996 revealed that rod-shaped bacteria were frequently associated with the small intestinal mucosa of CD patients, both in the active phase and in those treated with a GFD, as detected by scanning electron microscopy (SEM) related to increases in mRNA levels of mucin-2 and to alterations of the carbohydrate structures of the glycocalyx/mucous layer of the jejunal mucosa that could modify the specificity of bacterial adhesion [46] . However, further analysis of these samples by 16S rRNA gene sequencing and those obtained from a new cohort of patients (2004–2007) revealed that only one CD biopsy of the new cohort contained rod-shaped bacteria. The characterization of the microbiota from biopsies of CD patients from the Swedish CD epidemic showed that SEM-positive biopsies were significantly enriched in Prevotella spp. together with Clostridium and Actinomyces compared to the SEM-negative biopsies also from CD patients [43] , but the absence of such microbiome markers in other cohorts makes the theory inconsistent. By contrast, Nistal et al. [44] conducted a study with biopsies from CD children and adults with active and non-active disease and controls by 16S rRNA gene sequencing and reported that Prevotella spp. were reduced in the untreated versus treated group in adults and in the untreated versus control group in infants, suggesting a potential healthy role of this bacterial group. Finally, Cheng et al. [45] reported both increased and decreased abundance of Prevotella spp. depending on the species in biopsies of CD patients (P. melaninogenica increased) compared with controls (P. oralis increased).
Bifidobacterium and B. longum levels were found to be lower in the feces of CD patients with active and nonactive disease and in biopsies of CD patients with active disease compared to controls when analyzed by quantitative PCR [34]. An independent cultural study conducted with stool samples also found reduced Bifidobacterium species numbers but only in CD patients with active disease compared to treated CD patients and controls [37]. However, when the same research group conducted a comparison of biopsy and stool samples of children with non-active CD (under a GFD) and controls, differences in DGGE profiles of lactobacilli and bifidobacteria were found in biopsy samples as well as reduced bifidobacteria and other lactic acid bacterial counts in the stools of treated CD patients [39], confirming the same trend that abundance of bifidobacteria is reduced in CD. By DGGE, we also detected reduced bifidobacterial diversity in stools of active CD patients and reduced prevalence of B. adolescentis compared to controls, although non-active CD samples were not included [47]. By contrast, Nistal et al. [48] found reduced diversity of Bifidobacterium spp. in stools from CD subjects with non-active disease and increased prevalence of B. bifidum in CD patients with active disease by DGGE, which partly contradicts previous findings. All in all, most data indicated that reduced abundance of bifidobacteria is linked to CD, although the magnitude of the differences varies depending on the state of the disease and location in the intestinal tract (duodenal or fecal), likely as a consequence of other confounding factors not fully controlled for in previous studies (e.g. diet).
Enterobacteria and the corresponding phylum Proteobacteria has also been associated with CD but mainly with the active phase of the disease. An exception is the study by Schippa et al. [36] that reported an increased prevalence of Escherichia coli in duodenal biopsies from children with active and non-active CD compared to controls by TGGE. However, other studies suggest that enterobacteria numbers tend to be normalized after adherence to the GFD. Thus, E. coli numbers were found to be higher in feces and biopsies of nontreated CD patients than in those of controls, but not in patients under a GFD as determined by FISH and quantitative PCR [35, 40]. This trend was confirmed by cultural analysis of duodenal biopsies of CD patients with active and inactive disease and controls, where members of the phylum Proteobacteria (which include E. coli and other enterobacteria) were enriched in active CD while those of the phylum Firmicutes were less abundant [49]. The study by Wacklin et al. [33] included a low number of cases and only compared CD subjects with active disease and controls, but it also concluded that CD patients with gastrointestinal symptoms have increases in Proteobacteria in duodenal biopsies compared to controls and patients with dermatitis herpetiformis, using DGGE. This study also suggested that the microbiota may play a role in the manifestation of the disease. The same authors reported an increase in Proteobacteria in duodenal samples of patients under a GFD with persistent symptoms in support of the idea that this phylum is linked to CD symptoms [32]. Other cultural analyses using only stool samples of CD patients with active disease and controls confirmed this trend when analyzing total enterobacteria [38] or some representative groups ( Salmonella, Shigella and Klebsiella ) [39]. To date, there is only one study that characterized by virulence features of E. coli isolates from CD patients and controls, indicating that E. coli clones belonging to virulent phylogenetic groups (B2 and D) isolated from both untreated and treated CD patients presented a higher number of virulence genes, involved in adhesion and evasion of the host immune system (P fimbriae, capsule K5 and hemolysin) than those isolated from healthy controls [50]. These findings indicated that the enteric microbiota of CD subjects has a higher pathogenic potential than that of healthy subjects regardless of the state of the disease. It also suggested that more in-depth characterization of microbiota would be necessary to understand its role in the disease. The whole phylum Proteobacteria usually represents a minor component of the intestinal microbiota since these bacteria are facultative anaerobes, in contrast to most abundant phyla, which are obligated anaerobes [51]. The genetic variability and high frequency of conjugation-mediated horizontal gene transfer in enterobacteria might contribute to their fitness advantage over other members of the gut microbial community in particular conditions [51]. For example, commensal enterobacteria seem to be able to occupy an inflamed niche by acquiring a highly adhesive phenotype by pathoadaptive mutations resulting from persistent inflammation, as shown in Crohn’s disease [52]. The colonization of enterobacteria in a disease situation could also be favored by their ability to use nitrate generated from the inflammatory response of the host via anaerobic respiration, thereby outcompeting the obligate anaerobic bacteria [51].
Staphylococcus numbers have also been found to be higher in feces and biopsies of non-treated CD patients than in those of controls, but their levels were normalized after treatment with a GFD [35]. This trend was confirmed by one cultural study conducted with biopsies from the three groups of children [49] and two studies with stool samples but that only included untreated CD cases and controls [38, 39]. In the study by Sánchez et al. [49], members of the Staphylococcaceae family and, particularly, of the species S. epidermidis and S. pasteuri , were more abundant in patients with active disease than in controls. In contrast, members of the family Streptococcaceae were less abundant in patients with active CD than in controls. In biopsies from CD patients (children and adults), analysis by 16S rRNA gene sequencing also suggested a protective role for Streptococcus spp., but the sample size was very small [44]. Taking into account that streptococcus are normal inhabitants of the upper intestinal tract, the findings indicated that the disease is associated with the overgrowth of possible pathobionts (staphylococci) that exclude symbionts (streptococci) that are characteristic of the healthy small intestinal microbiota [49]. Nevertheless, more detailed analysis by isolation and identification of clones belonging to the genus Staphylococcus also revealed differences in the virulence features of the isolates from CD patients with active and non-active disease. In those CD patients, the species S. epidermidis carrying the mecA gene (methicillin-resistant gene) was more abundant than in controls [53].
There are only two studies revealing no differences in the total microbiome profile in small bowel biopsies of pediatric CD patients (untreated CD patients and controls) analyzed by means of IS-pro, a 16S-23S interspacer (IS) region-based profiling method [54], or mixing cases of children and adults with CD (treated and untreated) by quantitative PCR [55]; however, further analysis of the latter cases by microarrays revealed some differences [45].
The analysis of metabolites as indicators of intestinal dysbiosis (e.g. short-chain fatty acids, lactate, etc.) have also revealed some differences between CD patients, relatives and controls, but results are not consistent across studies, and decreases and increases of the same metabolite (e.g. butyrate) have been associated with CD [39, 56]. A more recent wide-spectrum metabolome analysis conducted by nuclear magnetic resonance found differences in urine metabolites that could be attributed to the CD microbiota, but most of the alterations were restored after adherence to the GFD [57] . Disease-associated alterations in microbiome-derived metabolites could not only reflect intestinal dysbiosis but also contribute to dysfunction of the immunoregulatory mechanisms. Thus, experimental models have proven that intestinal bacteria belonging to specific clostridial clusters or other commensals (such as Faecalibacterium prausnitzii) exert an immunoregulatory role by producing butyrate that, in turn, induces colonic Tregs via an epigenetic control of histone deacetylation [58, 59]. Nevertheless, so far, findings are not conclusive enough to understand the role of bacterial metabolites in CD pathogenesis and to what extent they reflect intestinal dysbiosis.
Disease-associated alterations in microbiome-derived metabolites could not only reflect intestinal dysbiosis but also contribute to dysfunction of the immunoregulatory mechanisms
So far, a few studies have analyzed the expression or production of molecules involved in host-microbe interactions as indirect evidence of intestinal dysbiosis as well as to identify the potential mechanism of action of the CD-associated intestinal microbiota. For example, increases in gene expression of α-defensins HD-5 and HD-6, which are the antimicrobial peptides produced by Paneth cells in response to bacterial antigens, have been reported in active CD but not in treated CD cases, supporting intestinal dysbiosis and probably a pathobiont expansion only in the active phase of the disease [31].
CD patients with active disease also showed increased sCD14 protein seropositivity, which was interpreted as a result of intestinal bacterial translocation resulting from gut barrier dysfunction that could be aggravated by intestinal dysbiosis. CD14 and TLR4 are involved in the recognition and signal transduction of the lipopolysaccharide (LPS), a major component of the bacterial cell wall of Gramnegative bacteria. The CD14/ TLR4 complex, upon binding, triggers innate host defense mechanisms, leading to the release of proinflammatory cytokines that could aggravate CD pathogenesis [60].
Alterations in TLR4 and TLR2 expression as well as functional single nucleotide polymorphism (SNP) in genes expressed upon TLR4 activation have also been reported to be associated with CD [31, 61]. As explained above, TLRs recognize bacterial or endogenous molecular patterns that, upon binding, trigger signal transduction via different adaptor molecules (MyD88 and TRIF) to induce proinflammatory gene expression. These play a vital role in host defense, but dysregulation in TLR signaling can also confer risk to autoimmune diseases, such as CD [61]. It is known that gut microbes provide signals that can both promote and inhibit autoimmunity by signaling through the TLR family [62]. While TRIF signaling could act as a negative regulator of immunity, MyD88 signaling seems to act as a potent activator. Indeed, ligands of TLRs that activate MyD88 signaling may be potent inducers of type-I IFN interferon production, with subsequent activation of the expression of other inflammatory inducible genes, leading to the production of cytokines such as IFN-α and TNF-α. Therefore, increases in the expression of some TLRs (such as TLR4) could contribute to amplifying the proinflammatory effects of intestinal dysbiosis in CD. Similarly, polymorphisms in genes which become active upon TLR4 signaling, such as those affecting IL-18R, could also influence the host-microbe cross talk in CD and contribute to mucosal inflammation [61]. In this context, several studies support that the relative proportion of Gram-negative to Gram-positive bacteria is increased in both CD patients (treated and untreated with a GFD) compared to controls [40, 41], and according to the more detailed analysis described above, the Gram-negative group could be represented by Bacteroides and Enterobacteriaceae, which could be a source of LPS for TLR4 activation.
Simultaneous microbiome and host signaling pathways and inflammatory marker analyses that help linking symbionts or pathobionts with CD have only been reported in studies including a small number of CD cases [45, 55]. The findings showed a decreased gene expression of TLR2 and TOLLIP (a negative regulator of TLR signaling) and increased expression of TLR9 in small intestinal biopsies of CD patients. Also, an increased expression of IL-10 and IFN-α was found in CD and suggested to be mediated by microbiota-associated factors via TLR9 signaling [45, 55]. However, only minor differences in eight genus-like bacteria were reported between CD and healthy subjects by the use of microarrays [45, 55], which was not confirmed in studies by other authors.
Increases in the expression of some TLRs could contribute to amplifying the proinflammatory effects of intestinal dysbiosis in CD
In all these studies conducted with CD patients, the GFD per se may cause changes in the intestinal microbiota composition beyond the underlying disease, which acts as inevitable confounding factor [63] . This was demonstrated in healthy adults, in whom the adherence to a GFD led to reductions in the intake of complex polysaccharides and caused parallel shifts in gut microbiota composition, reducing the numbers of bifidobacteria and increasing those of enterobacteria [63] . This makes it particularly difficult to dissect the cause-consequence cycle and establish whether intestinal dysbiosis could promote CD onset. To do so, prospective studies in healthy infants at family risk of CD are underway [12, 14] . These studies have revealed that the HLA-DQ genotype, in itself, influences the intestinal microbiota composition. Infants with a high-risk genotype (HLA-DQ2 genotype) showed an increased proportion of Firmicutes and Proteobacteria and a reduction in Actinobacteria (including the genus Bifidobacterium ) compared to controls (no HLA-DQ genotype). These findings suggest that the CD host genotype selects for the early colonizers of the infant’s gut, which together with environmental factors, such as the type of milk feeding, could influence the development of oral tolerance to gluten [14] . However, this hypothesis should be confirmed by proving that this altered bacterial colonization pattern is linked to CD onset.
Therefore, there is a need to progress in the identification of the type of receptors and signaling pathways involved in microbiota-mediated aggravation of CD and to understand whether CD patients or predisposed individuals have a compromised ability to acquire and maintain a symbiotic microbiota, increasing the fitness of pathobionts that can promote CD onset. Investigation should also advance on the identification of specific strain pathogenic features that can contribute to the breakdown of the peaceful host-microbiome dynamics in CD in larger and multidisciplinary studies.
Role of Intestinal Dysbiosis in CD Pathogenesis: Lessons from Experimental Models
Gnotobiotic animal models provide a useful experimental tool to investigate causality based on clinical associations of altered intestinal microbiota composition and function in CD patients [64]. A recent study in mice expressing the human HLA-DQ8 gene, which provides a moderate genetic risk to CD, suggests that the microbiota may be a modulator of host immune responses to gluten [65]. NOD-DQ8 mice we raised in different microbial background conditions, which included strict germ-free conditions, colonized with a benign microbiota composed of 8 bacterial strains [the altered Schaedler flora (ASF)], or under conventional conditions [specific pathogen free (SPF)]. The main difference between the ASF and SPF colonized states was the presence of a limited micro biota with anti-inflammatory characteristics (Lactobacillus spp. and Parabacteroides distasonis) in ASF mice versus a complex microbiota with presence of potential pathobionts (Proteobacteria) in SPF mice. The study showed that the microbiota background had a significant effect on the degree of host immune responses to gluten. In the absence of commensal colonization (germ-free status) or in the presence of pathobionts (SPF), gluten-induced immunopathology was more severe than in mice that were colonized with anti-inflammatory bacteria (ASF). Moreover, the addition of a pathobiont, E. coli ENT CAI:5, isolated from CD patients [50] to the mice colonized with protective bacteria, reversed the protective effect and induced immunopathology in the presence of gluten sensitization [65].
Functional in vivo studies are needed to understand the pathogenic role of specific pathobionts in the altered response to gluten
Further evidence of the potential proinflammatory role of intestinal dysbiosis found in CD was demonstrated by exposure of peripheral blood mononuclear cell cultures to stool samples of patients with active and non-active CD, which led to an increased production of proinflammatory cytokine secretion (IFN-γ and TNF-α) compared to the stools of healthy subjects. In the same experimental setup, bifidobacteria ( B. longum CECT 7347 and B. bifidum CECT 7365) reduced the inflammatory cytokine secretion (IFN-γ and TNF-α) induced by the fecal microbiota of CD patients while increasing IL-10 secretion [31] . Further studies also demonstrated that enterobacteria isolated from CD patients could aggravate the effect of CD triggers (gliadin) in germ-free rat intestinal loops and in vitro cell cultures [31]. The adverse effects include: (i) reduction of the gut barrier function reflected in reductions in the number of mucus-producing cells and alterations in tightjunction protein expression, which facilitated gliadin translocation to the lamina propria; (ii) enhanced secretion of inflammatory mediators, and (iii) reduction of the TIMP metallopeptidase inhibitor 1 release, which could lead to an increase in activity of the matrix metalloproteinases involved in degrading the extracellular matrix and tissue damage in the intestinal loop. In cell cultures, enterobacteria isolated from CD patients also induced marked alterations on morphological and functional features of monocyte-derived DCs, such as podosome dissolution and dendrites, and activation of DC adhesion and spreading as well as inflammatory cytokine secretion (IFN-γ, TNF-α, IL-12) by DCs and peripheral blood mononuclear cells, partially resembling the gliadin-induced Th1-type cytokine profile [66]. Comparative studies were done with bifidobacteria showing no effect or an amelioration of gliadin and enterobacteria-induced effects [31, 66]. Nevertheless, findings from in vitro studies are of limited value, and further functional in vivo studies are needed to understand the pathogenic role of specific pathobionts in the altered response to gluten.
Translational and Clinical Implications
An unprecedented number of studies have been conducted to identify the features of a ‘healthy microbiome/ microbiota’ and the alterations on the host-microbiota cross talk, promoting the progress from health to disease in diverse disorders. Nevertheless, this is still a major challenge due to the complexity of the microbiota, the redundancy of its functions and its dependency on life events that are influenced by environmental and individual genetic factors. Furthermore, disorders in which the microbiota is thought to play a role have a multifactorial etiology and show a wide spectrum of manifestations that could be related to the underlying etiologic mechanisms, which complicate the scene.
Advances in this area could, however, open the way to the development of microbiome-informed strategies for stratified and personalized preventive and therapeutic measures for immune-mediated disorders such as CD. These could be based on dietary strategies that help optimize the partnership between the gut microbiota and host immunity, increasing its stability and resilience to disease. Furthermore, the identification of the host pathways modulated by the microbiota and their agonists could provide feasible approaches to improve disease management.
Acknowledgements
The work was supported by grant AGL2011-25169 from the Spanish Ministry of Economy and Competitiveness (MINECO). I also thank Dr. E.F. Verdu from McMaster University for the scientific discussion related to this paper and critical review.
Disclosure Statement
Y. Sanz developed a patent belonging to CSIC on a probiotic strain for CD, but the patent was licensed and she did not receive any personal funding. The writing of this article was supported by Nestlé Nutrition Institute.
References
Gerritsen J, Smidt H, Rijkers GT, de Vos WM: Intestinal microbiota in human health and disease: the impact of probiotics. Genes Nutr 2011; 6: 209–240.
Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE: Metagenomic analysis of the human distal gut microbiome. Science 2006; 312: 1355– 1359.
Hooper LV, Littman DR, Macpherson AJ: Interactions between the microbiota and the immune system. Science 2012; 336: 1268– 1273.
Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, van den Brandt PA, Stobberingh EE: Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006; 118: 511–521.
Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, Khan MT, Zhang J, Li J, Xiao L, Al- Aama J, Zhang D, Lee YS, Kotowska D, Colding C, Tremaroli V, Yin Y, Bergman S, Xu X, Madsen L, Kristiansen K, Dahlgren J, Wang J: Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 2015; 17: 690–703.
Pozo-Rubio T, Olivares M, Nova E, De Palma G, Mujico JR, Ferrer MD, Marcos A, Sanz Y: Immune development and intestinal microbiota in celiac disease. Clin Dev Immunol 2012; 2012: 654143.
Sanz Y, De Palma G: Gut microbiota and probiotics in modulation of epithelium and gutassociated lymphoid tissue function. Int Rev Immunol 2009; 28: 397–413.
Tlaskalová-Hogenová H, Stěpánková R, Kozáková H, Hudcovic T, Vannucci L, Tučková L, Rossmann P, Hrnčíř T, Kverka M, Zákostelská Z, Klimešová K, Přibylová J, Bártová J, Sanchez D, Fundová P, Borovská D, Srůtková D, Zídek Z, Schwarzer M, Drastich P, Funda DP: The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol 2011; 8: 110–120.
Alexander KL, Targan SR, Elson CO 3rd: Microbiota activation and regulation of innate and adaptive immunity. Immunol Rev 2014; 260: 206–220.
Sartor RB: The intestinal microbiota in inflammatory bowel diseases. Nestle Nutr Inst Workshop Ser 2014; 79: 29–39.
Cenit MC, Olivares M, Codoñer-Franch P, Sanz Y: Intestinal microbiota and celiac disease: cause, consequence or co-evolution? Nutrients 2015; 7: 6900–6923.
Zhang X, Zhang D, Jia H, et al: The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 2015; 21: 895–905.
Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A, Palau F, Nova E, Marcos A, Polanco I, Ribes-Koninckx C, Ortigosa L, Izquierdo L, Sanz Y: The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut 2015; 64: 406–417.
Han A, Newell EW, Glanville J, Fernandez- Becker N, Khosla C, Chien YH, Davis MM: Dietary gluten triggers concomitant activation of CD4+ and CD8+ αβ T cells and γδ T cells in celiac disease. Proc Natl Acad Sci USA 2013; 110: 13073–13078.
Kim SM, Mayassi T, Jabri B: Innate immunity: actuating the gears of celiac disease pathogenesis. Best Pract Res Clin Gastroenterol 2015; 29: 425–435.
Palová-Jelínková L, Dáňová K, Drašarová H, Dvořák M, Funda DP, Fundová P, Kotrbová- Kozak A, Černá M, Kamanová J, Martin SF, Freudenberg M, Tučková L: Pepsin digest of wheat gliadin fraction increases production of IL-1β via TLR4/MyD88/TRIF/MAPK/ NF-κB signaling pathway and an NLRP3 inflammasome activation. PLoS One 2013; 8:e62426.
Caruso R, Marafini I, Sedda S, Del Vecchio Blanco G, Giuffrida P, MacDonald TT, Corazza GR, Pallone F, Di Sabatino A, Monteleone G: Analysis of the cytokine profile in the duodenal mucosa of refractory coeliac disease patients. Clin Sci (Lond) 2014; 126: 451–458.
Thomas KE, Sapone A, Fasano A, Vogel SN: Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: role of the innate immune response in celiac disease. J Immunol 2006; 176: 2512–2521.
Junker Y, Zeissig S, Kim SJ, Barisani D, Wieser H, Leffler DA, Zevallos V, Libermann TA, Dillon S, Freitag TL, Kelly CP, Schuppan D: Wheat amylase trypsin inhibitors drive intestinal inflammation via activation of toll-like receptor 4. J Exp Med 2012; 209: 2395–2408.
Marafini I, Monteleone I, Di Fusco D, Cupi ML, Paoluzi OA, Colantoni A, Ortenzi A, Izzo R, Vita S, De Luca E, Sica G, Pallone F, Monteleone G: TNF-α producing innate lymphoid cells (ILCs) are increased in active celiac disease and contribute to promote intestinal atrophy in mice. PLoS One 2015; 10: e0126291.
Araya RE, Jury J, Bondar C, Verdu EF, Chirdo FG: Intraluminal administration of poly I:C causes an enteropathy that is exacerbated by administration of oral dietary antigen. PLoS One 2014; 9:e99236.
Abed J, Lebreton C, Champier G, Cuvillier A, Cogné M, Meresse B, Dugave C, Garfa- Traoré M, Corthésy B, Cerf-Bensussan N, Heyman M: Abnormal apical-to-basal transport of dietary ovalbumin by secretory IgA stimulates a mucosal Th1 response. Mucosal Immunol 2014; 7: 315–324.
Fasano A: Zonulin, regulation of tight junctions, and autoimmune diseases. Ann NY Acad Sci 2012; 1258: 25–33.
van Leeuwen MA, Lindenbergh-Kortleve DJ, Raatgeep HC, de Ruiter LF, de Krijger RR, Groeneweg M, Escher JC, Samsom JN: Increased production of interleukin-21, but not interleukin-17A, in the small intestine characterizes pediatric celiac disease. Mucosal Immunol 2013; 6: 1202–1213.
Park SJ, Shin JI: Interleukin-21-mediated TLR4 activation in celiac disease. Dig Dis Sci 2013; 58: 2425.
Panea C, Farkas AM, Goto Y, Abdollahi- Roodsaz S, Lee C, Koscsó B, Gowda K, Hohl TM, Bogunovic M, Ivanov II: Intestinal monocyte-derived macrophages control commensal-specific Th17 responses. Cell Rep 2015; 12: 1314–1324.
Cicerone C, Nenna R, Pontone S: Th17, intestinal microbiota and the abnormal immune response in the pathogenesis of celiac disease. Gastroenterol Hepatol Bed Bench 2015; 8: 117–122.
Sarra M, Cupi ML, Monteleone I, Franzè E, Ronchetti G, Di Sabatino A, Gentileschi P, Franceschilli L, Sileri P, Sica G, Del Vecchio Blanco G, Cretella M, Paoluzi OA, Corazza GR, Pallone F, Monteleone G: IL-15 positively regulates IL-21 production in celiac disease mucosa. Mucosal Immunol 2013; 6: 244– 255.
Stepniak D, Koning F: Celiac disease – sandwiched between innate and adaptive immunity. Hum Immunol 2006; 67: 460–468.
Sanz Y, De Pama G, Laparra M: Unraveling the ties between celiac disease and intestinal microbiota. Int Rev Immunol 2011; 30: 207– 218.
Wacklin P, Laurikka P, Lindfors K, Collin P, Salmi T, Lähdeaho ML, Saavalainen P, Mäki M, Mättö J, Kurppa K, Kaukinen K: Altered duodenal microbiota composition in celiac disease patients suffering from persistent symptoms on a long-term gluten-free diet. Am J Gastroenterol 2014; 109: 1933–1941.
Wacklin P, Kaukinen K, Tuovinen E, Collin P, Lindfors K, Partanen J, et al: The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm Bowel Dis 2013; 19: 934–941.
Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y: Imbalances in faecal and duodenal Bifidobacterium species composition in active and non-active coeliac disease. BMC Microbiol 2008; 8: 232.
Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y: Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J Clin Pathol 2009; 62: 264–269.
Schippa S, Iebba V, Barbato M, Di Nardo G, Totino V, Checchi MP, et al: A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol 2010; 10: 175.
Di Cagno R, Rizzello CG, Gagliardi F, Ricciuti P, Ndagijimana M, Francavilla R, Guerzoni ME, Crecchio C, Gobbetti M, De Angelis M: Different fecal microbiotas and volatile organic compounds in treated and untreated children with celiac disease. Appl Environ Microbiol 2009; 75: 3963–3971.
Collado MC, Calabuig M, Sanz Y: Differences between the fecal microbiota of coeliac infants and healthy controls. Curr Issues Intest Microbiol 2007; 8: 9–14.
Di Cagno R, De Angelis M, De Pasquale I, Ndagijimana M, Vernocchi P, Ricciuti P, Gagliardi F, Laghi L, Crecchio C, Guerzoni ME, Gobbetti M, Francavilla R: Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol 2011; 11: 219.
Nadal I, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y: Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J Med Microbiol 2007; 56: 1669–1674.
De Palma G, Nadal I, Medina M, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y: Intestinal dysbiosis and reduced immunoglobulin- coated bacteria associated with coeliac disease in children. BMC Microbiol 2010; 10: 63.
Sánchez E, Laparra JM, Sanz Y: Discerning the role of Bacteroides fragilis in celiac disease pathogenesis. Appl Environ Microbiol 2012; 78:6507–6515.
Ou G, Hedberg M, Hörstedt P, Baranov V, Forsberg G, Drobni M, et al: Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. Am J Gastroenterol 2009; 104: 3058–3067.
Nistal E, Caminero A, Herrán AR, Arias L, Vivas S, de Morales JM, Calleja S, de Miera LE, Arroyo P, Casqueiro J: Differences of small intestinal bacteria populations in adults and children with/without celiac disease: effect of age, gluten diet, and disease. Inflamm Bowel Dis 2012; 18: 649–656.
Cheng J, Kalliomäki M, Heilig HG, Palva A, Lähteenoja H, de Vos WM, Salojärvi J, Satokari R: Duodenal microbiota composition and mucosal homeostasis in pediatric celiac disease. BMC Gastroenterol 2013; 13: 113.
Forsberg G, Fahlgren A, Hörstedt P, Hammarström S, Hernell O, Hammarström ML: Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am J Gastroenterol 2004; 99: 894–904.
Sanz Y, Sánchez E, Marzotto M, Calabuig M, Torriani S, Dellaglio F: Differences in faecal bacterial communities in coeliac and healthy children as detected by PCR and denaturing gradient gel electrophoresis. FEMS Immunol Med Microbiol 2007; 51: 562–568.
Nistal E, Caminero A, Vivas S, Ruiz de Morales JM, Sáenz de Miera LE, Rodríguez- Aparicio LB, Casqueiro J: Differences in faecal bacteria populations and faecal bacteria metabolism in healthy adults and celiac disease patients. Biochimie 2012; 94: 1724–1729.
Sánchez E, Donat E, Ribes-Koninckx C, Fernández-Murga ML, Sanz Y: Duodenalmucosal bacteria associated with celiac disease in children. Appl Environ Microbiol 2013; 79: 5472–5479.
Sánchez E, Nadal I, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y: Reduced diversity and increased virulence-gene carriage in intestinal enterobacteria of coeliac children. BMC Gastroenterol 2008; 8: 50.
Shin NR, Whon TW, Bae JW: Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 2015; 33: 496– 503.
Iebba V, Conte MP, Lepanto MS, Di Nardo G, Santangelo F, Aloi M, Totino V, Checchi MP, Longhi C, Cucchiara S, Schippa S: Microevolution in fimH gene of mucosa-associated Escherichia coli strains isolated from pediatric patients with inflammatory bowel disease. Infect Immun 2012; 80: 1408–1417.
Sánchez E, Ribes-Koninckx C, Calabuig M, Sanz Y: Intestinal Staphylococcus spp. and virulent features associated with coeliac disease. J Clin Pathol 2012; 65: 830–834.
de Meij TG, Budding AE, Grasman ME, Kneepkens CM, Savelkoul PH, Mearin ML: Composition and diversity of the duodenal mucosa-associated microbiome in children with untreated coeliac disease. Scand J Gastroenterol 2013; 48: 530–536.
Kalliomäki M, Satokari R, Lähteenoja H, Vähämiko S, Grönlund J, Routi T, Salminen S: Expression of microbiota, Toll-like receptors, and their regulators in the small intestinal mucosa in celiac disease. J Pediatr Gastroenterol Nutr 2012; 54: 727–732.
Tjellström B, Stenhammar L, Högberg L, Fälth-Magnusson K, Magnusson KE, Midtvedt T, Sundqvist T, Houlston R, Popat S, Norin E: Gut microflora associated characteristics in first-degree relatives of children with celiac disease. Scand J Gastroenterol 2007; 42: 1204–1208.
Bertini I, Calabrò A, De Carli V, Luchinat C, Nepi S, Porfirio B, Renzi D, Saccenti E, Tenori L: The metabonomic signature of celiac disease. J Proteome Res 2009; 8: 170–177.
Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K: Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011; 331: 337–341.
Qiu X, Zhang M, Yang X, Hong N, Yu C: Faecalibacterium prausnitzii upregulates regulatory T cells and anti-inflammatory cytokines in treating TNBS-induced colitis. J Crohns Colitis 2013; 7:e558–e568.
Hoffmanová I, Sánchez D, Hábová V, Anděl M, Tučková L, Tlaskalová-Hogenová H: Serological markers of enterocyte damage and apoptosis in patients with celiac disease, autoimmune diabetes mellitus and diabetes mellitus type 2. Physiol Res 2015; 64: 537– 546.
Kim S, Becker J, Bechheim M, Kaiser V, Noursadeghi M, Fricker N, Beier E, Klaschik S, Boor P, Hess T, Hofmann A, Holdenrieder S, Wendland JR, Fröhlich H, Hartmann G, Nöthen MM, Müller-Myhsok B, Pütz B, Hornung V, Schumacher J: Characterizing the genetic basis of innate immune response in TLR4-activated human monocytes. Nat Commun 2014; 5: 5236.
Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV: Microbiota regulates type 1 diabetes through Toll-like receptors. Proc Natl Acad Sci USA 2015; 112: 9973–9977.
De Palma G, Nadal I, Collado MC, Sanz Y: Effects of a gluten-free diet on gut microbiota and immune function in healthy adult human subjects. Br J Nutr 2009; 102: 1154–1160.
Verdu EF, Galipeau HJ, Jabri B: Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol 2015; 12: 497–506.
Galipeau HJ, McCarville JL, Huebener S, Litwin O, Meise M, Jabri B, Sanz Y, Murray JA, Jordana M, Alaedini A, Chirdo FG, Verdu EF: Microbiota modulates the response to dietary gluten in gnotobiotic mice. Am J Pathol 2015, in press.
De Palma G, Kamanova J, Cinova J, Olivares M, Drasarova H, Tuckova L, Sanz Y: Modulation of phenotypic and functional maturation of dendritic cells by intestinal bacteria and gliadin: relevance for celiac disease. J Leukoc Biol 2012; 92: 1043–1054.